TMA
Laboratorio > ► Ensayos Termicos
Análisis termomecánico (TMA)
El análisis termomecánico (TMA) mide la estabilidad de la forma de un material a temperaturas elevadas al penetrarlo físicamente con una varilla de metal. En TMA, la temperatura de la muestra de prueba se eleva a una velocidad constante, la muestra se coloca dentro del dispositivo de medición y se coloca encima una varilla con un peso específico. Para permitir las mediciones a bajas temperaturas, la muestra, el horno y la varilla se pueden enfriar con nitrógeno líquido. La mayoría de los instrumentos son tan precisos que se pueden utilizar para medir la temperatura de fusión del material y, mediante dilatometría lineal, para medir los coeficientes de expansión térmica.
TMA
El análisis termomecánico (TMA) es una técnica termoanalítica establecida basada en la medición de cambios en la longitud de la muestra (L) o el volumen (V) en función de la temperatura o el tiempo, bajo carga a presión atmosférica. La técnica también se conoce como termodilatometría (TD) si las dimensiones se miden con una fuerza insignificante que actúa sobre el material de prueba mientras se somete a un programa de temperatura controlada. Los estudios termomecánicos se realizan generalmente bajo carga estática con una variedad de sondas para medir cambios dimensionales en los modos de prueba de expansión/compresión, penetración, tensión, flexión o cizallamiento. Mediante la aplicación de modos especiales y diferentes aditamentos, también son factibles las mediciones de relajación de esfuerzos, fluencia, reometría de placas paralelas y dilatometría de volumen. Los materiales poliméricos se suelen estudiar en forma de sólidos rígidos o casi rígidos, o en estado líquido (fundido) utilizando accesorios especiales. También se pueden utilizar para medir los cambios de volumen en muestras de forma irregular o polvos sumergidos en un líquido inerte (por ejemplo, en dilatómetros de mercurio). Las cantidades físicas básicas utilizadas en TMA son la tensión, σ, que se define como la fuerza aplicada por unidad de área del material, σ=F/A, con A denotando el área de la sección transversal de la muestra, y la deformación por unidad de dimensión causada por la tensión aplicada y medida por la deformación, ε. Para un experimento de tracción simple, la deformación se define como ε = ΔL/L, donde ΔL es el cambio de longitud y L es la longitud original. Cuando se somete a una fuerza mecánica, los materiales pueden comportarse de diversas formas. Un material quebradizo se deformará reversiblemente a una pequeña cantidad y luego se fracturará, mientras que un material dúctil también se deformará reversiblemente hasta una cierta cantidad y luego cederá y fluirá bajo la fuerza aplicada hasta que comience a endurecerse bajo carga y luego fallar. Hasta el límite elástico, el material volverá a su forma y tamaño anteriores cuando se elimine la fuerza. La pendiente de esta región lineal corresponde al módulo de Young, E, también conocido como módulo elástico, módulo de tracción o simplemente módulo. Durante las mediciones de expansión de polímeros amorfos y semicristalinos que no están orientados, se observa un aumento repentino en la velocidad de expansión por encima de Tg, a medida que el material cambia de una configuración estructural de movilidad de cadena limitada o nula a un estado de movilidad de cadena aumentada. El punto de intersección, visto como una inflexión o doblez, de las curvas de expansión de volumen o lineal vítrea y gomosa define típicamente la temperatura de transición vítrea. Sin embargo, tenga en cuenta que se observa un valor de Tg algo diferente para cada modo de prueba, ya que cada uno mide un efecto diferente. El cambio en la pendiente de las curvas de expansión por debajo y por encima de Tg está relacionado con la expansión del volumen libre, ya que el volumen real de las moléculas (el volumen incondicional o incompresible, correspondiente a temperatura termodinámica cero o presión extremadamente alta) no cambia apreciablemente alrededor de Tg. Solo en el caso de sistemas semicristalinos, un aumento adicional de la temperatura puede provocar la penetración de la sonda en la muestra, incluso con una carga insignificante sobre ella. Esta abrupta disminución en la posición de la sonda (temperatura de inicio), en la mayoría de los casos puede asignarse a la fusión del cristal, y la temperatura de la ruptura en la curva representa el punto de fusión.
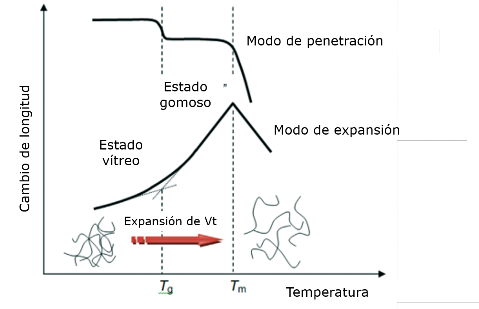
Idealmente, la expansión térmica lineal y la posterior contracción de una muestra a sus dimensiones originales son fenómenos totalmente reversibles. No obstante, si el material se ablanda a medida que se calienta mientras se somete a una carga mecánica, fluirá y se deslizará, lo que provocará una deformación dimensional irreversible.
Además, si el material se estiró (orientó) cuando estaba blando y luego se enfrió antes del experimento, las tensiones residuales permanecerán en la muestra (efectos de memoria). Dentro de las condiciones del experimento termomecánico, la relajación de la tensión en el calentamiento provocará cambios morfológicos irreversibles (es decir, aleatorización de la orientación lograda durante la fabricación) junto con la contracción del material de prueba. Los cambios de longitud típicamente medidos por TMA convencional son, por tanto, una convolución de los efectos anteriores, a menos que la muestra sea completamente isótropa y las mediciones se realicen bajo cargas cero. Se logra una separación de cambios dimensionales superpuestos termodinámicos y cinéticos, reversibles y no reversibles en experimentos de TMA de temperatura modulada (MTTMA). En este caso, la cantidad física dependiente medida es la longitud, con la muestra expuesta a una modulación de temperatura sinusoidal superpuesta a un perfil lineal subyacente de calentamiento, enfriamiento o isotermo, similar al MTDSC. Las condiciones de modulación son diferentes de MTDSC, ya que la muestra y el dispositivo de prueba y el recinto son más grandes en la prueba mecánica, por lo que requieren tiempos de equilibrio (y escaneo) más largos. Varios estudios demuestran la usabilidad de TMA como una técnica complementaria a DSC, DTA o estudios de análisis termogravimétrico, en la realización de caracterización estructural y térmica de sistemas poliméricos homogéneos (por ejemplo, mezclas de poli (vinil fenil cetona hidrogenada (PVPhKH) con poli (2etil-2-oxazolina) o poli (estireno-co-4-vinilpiridina) (PS4VP)), mezclas que muestran una elevada heterogeneidad estructural en composiciones de mezcla seleccionadas (p. Ej., Mezclas de PVPhKH + PPO), o fase -sistemas separados (poliuretano + PMMA, PVC + PMMA.